
Charge Propagation Workflow
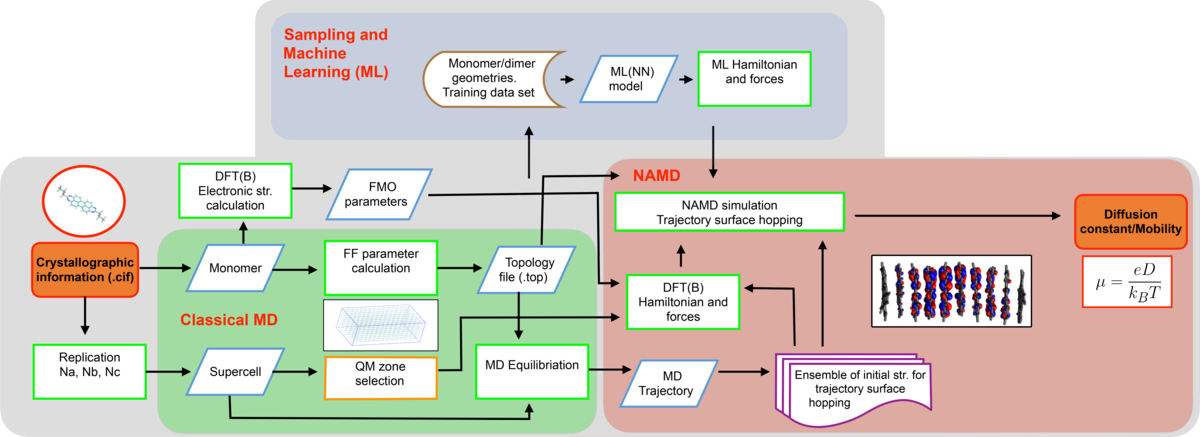
The calculation of intrinsic charge mobilities in organic semiconductors is a complex, multifaceted process. It involves several critical steps, including classical molecular dynamics (MD) simulations for system preparation, manual computation of reorganization energy, non-adiabatic simulations to evaluate transfer integrals, followed by production runs and simulations to ultimately determine charge mobilities.
To address these challenges, a workflow is under development to automate this entire process. The objective of this software is to simplify the complexities associated with the simulation chain, facilitating the calculation of key properties essential for assessing the viability of newly discovered organic semiconductors. To carry out these calculations, the crystal structure of the molecule to be analyzed is required as the foundation for the simulations. Additionally, the system is designed to handle large datasets, enabling the calculation of charge mobilities for numerous molecules to support the training of neural networks.
| Name | Position | Portrait | |
|---|---|---|---|
| Sebastian Will | Ph.D. Student |